Vertex Announces Positive Results From the VX-548 Phase 3 Program for the Treatment of Moderate-to-Severe Acute Pain
– Treatment with VX-548 led to statistically significant improvement in pain compared to placebo as well as a clinically meaningful reduction in pain from baseline in both the abdominoplasty and bunionectomy randomized controlled trials –
– Treatment with VX-548 was also shown to be effective in the single arm study in a broad range of surgical and non-surgical pain conditions for up to 14 days –
– VX-548 was safe and well tolerated in all three studies –
– Vertex plans to submit a New Drug Application to the FDA by mid-2024 –
– Vertex to host investor call January 30 at 8:00 a.m. ET –
Vertex Pharmaceuticals Incorporated (NASDAQ:VRTX) today announced positive results from its Phase 3 program for the selective NaV1.8 inhibitor, VX-548, in the treatment of moderate-to-severe acute pain. The Phase 3 program included two randomized, double-blind, placebo-controlled, pivotal trials, one following abdominoplasty surgery and one following bunionectomy surgery, as well as a single arm safety and effectiveness study which enrolled patients with a broad range of surgical and non-surgical pain conditions.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20240129843577/en/
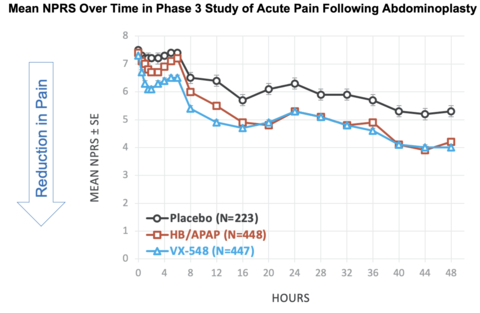
Figure 1: Mean NPRS Over Time in Phase 3 Study of Acute Pain Following Abdominoplasty (Graphic: Business Wire)
Treatment with VX-548 following abdominoplasty or bunionectomy surgery resulted in a statistically significant improvement on the primary endpoint of the time-weighted sum of the pain intensity difference from 0 to 48 hours (SPID48) compared to placebo as well as a clinically meaningful reduction in pain from baseline at 48 hours on the Numeric Pain Rating Scale (NPRS) in both studies (abdominoplasty: LS mean difference in SPID48 between VX-548 and placebo = 48.4 (95% CI: 33.6, 63.1; P<0.0001); bunionectomy: LS mean difference in SPID48 between VX-548 and placebo = 29.3 (95% CI: 14.0, 44.6; P=0.0002)).
For the first key secondary endpoint, Vertex tested the hypothesis that VX-548 was superior to hydrocodone bitartrate/acetaminophen (HB/APAP) on SPID48 following abdominoplasty surgery or bunionectomy surgery. Neither trial met this key secondary endpoint (abdominoplasty: LS mean difference between VX-548 and HB/APAP = 6.6 (95% CI: -5.4, 18.7; P=0.2781); bunionectomy: LS mean difference between VX-548 and HB/APAP = -20.2 (95% CI: -32.7, -7.7; P=0.0016)).
The second key secondary endpoint in both trials was time to meaningful pain relief defined as ≥2-point reduction in NPRS from baseline compared to placebo. VX-548 had a more rapid onset to meaningful pain relief than placebo in both the abdominoplasty and bunionectomy trials. (The median time to meaningful pain relief was 8 hours for placebo in both studies compared to 2 hours in abdominoplasty and 4 hours in bunionectomy for VX-548, with nominal P<0.0001 and 0.0016, respectively.)
Other secondary endpoints in both trials were generally consistent with the primary endpoint.
The Phase 3 single arm safety and effectiveness study evaluated treatment with VX-548 for up to 14 days across a broad range of other surgical and non-surgical acute pain conditions and demonstrated favorable safety and tolerability, as well as effectiveness as measured by a Patient Global Assessment (PGA) at the end of treatment (83.2% of patients rated VX-548 as good, very good, or excellent in treating pain).
VX-548 was safe and well tolerated in all three Phase 3 studies. The majority of adverse events (AEs) were mild to moderate, and there were no serious adverse events (SAEs) related to VX-548. In general, AEs in the two randomized controlled trials were consistent with the post-surgical setting. In the VX-548 arm, the incidence of AEs was lower than placebo (patients with any AEs in VX-548 and placebo arms: 50.0% and 56.3%, respectively, following abdominoplasty, and 31.0% and 35.2%, respectively, following bunionectomy).
"We are very pleased with the results from the VX-548 pivotal program, which demonstrate a compelling and consistent combination of efficacy and safety across multiple acute pain conditions and settings. The VX-548 benefit-risk profile ideally positions it to potentially fill the gap between medicines with good tolerability but limited efficacy and opioid medicines with therapeutic efficacy but known risks, including addictive potential," said Reshma Kewalramani, M.D., Chief Executive Officer and President of Vertex. "With FDA Breakthrough and Fast Track Designations in hand, we are working with urgency to file the New Drug Application for VX-548 and bring this non-opioid medicine to the millions of patients who suffer from acute pain each year in the U.S."
"As a physician treating patients suffering from pain for many years, I know firsthand the critical need for new, efficacious and safe treatment options," said Jessica Oswald, M.D., M.P.H., Associate Physician in Emergency Medicine and Pain Medicine, University of California San Diego, and Vertex Acute Pain Steering Committee Member. "The Phase 3 safety and efficacy across the three studies are impressive and demonstrate VX-548's potential to change the paradigm of pain management. I look forward to the potential of having a new class of acute pain medicine — the first in more than two decades — to use as an alternative to opioids to help the millions of people impacted by acute pain."
VX-548 Phase 3 Results in Patients Undergoing Abdominoplasty
Efficacy Results
Patients aged 18 to 80 years with moderate or severe pain after abdominoplasty surgery were eligible to participate in the trial. 1,118 patients were randomized and dosed with either VX-548 administered orally with an initial dose of 100 mg followed by 50 mg every 12 hours (at 12, 24 and 36 hours after the first dose), hydrocodone bitartrate/acetaminophen (5 mg/325 mg administered orally every 6 hours over 42 hours), or placebo.
Primary and Key Secondary Outcomes in Phase 3 Study of Acute Pain Following Abdominoplasty
Placebo
|
HB/APAP
|
VX-548
|
|
Primary endpoint of SPID48 vs. placebo |
|
|
|
LS mean SPID48 (SE) |
70.1 (6.1) |
-- |
118.4 (4.3) |
LS mean SPID48 difference from placebo |
-- |
-- |
48.4 |
95% CI |
-- |
-- |
(33.6, 63.1) |
P value vs. placebo |
-- |
-- |
<0.0001 |
First key secondary endpoint of SPID48 vs. HB/APAP |
|
|
|
LS mean SPID48 (SE) |
-- |
111.8 (4.3) |
118.4 (4.3) |
LS mean SPID48 difference from HB/APAP |
-- |
-- |
6.6 |
95% CI |
-- |
-- |
(-5.4, 18.7) |
P value vs. HB/APAP |
-- |
-- |
0.2781 |
Second key secondary endpoint of time to ≥2-point reduction in NPRS from baseline vs. placebo (minutes) |
|
|
|
Median time |
480 |
-- |
119 |
95% CI |
(477, 705) |
-- |
(90, 180) |
P value* vs. placebo (Log-rank) |
-- |
-- |
<0.0001 |
*P value is nominal; CI = confidence interval; LS = least squares; SE = standard error. |
|||
Mean NPRS Over Time in Phase 3 Study of Acute Pain Following Abdominoplasty [See Figure #1 at top of press release]
NPRS Reductions From Baseline at 48 Hours in Phase 3 Study of Acute Pain Following Abdominoplasty
Placebo
|
HB/APAP
|
VX-548
|
||
Baseline NPRS, mean |
7.5 |
7.4 |
7.3 |
|
Change from baseline in NPRS at 48 hours, mean |
-2.3 |
-3.2 |
-3.4 |
|
% reduction from baseline in mean NPRS at 48 hours |
31% |
43% |
47% |
Safety Results
VX-548 was generally well tolerated in this study. The majority of adverse events (AEs) were mild to moderate, and there were no serious adverse events (SAEs) related to VX-548.
In general, AEs were consistent with the post-surgical setting. In the VX-548 arm, the incidence of AEs was lower than placebo (patients with any AEs in VX-548 and placebo arms: 50.0% and 56.3%, respectively).
AEs with an incidence ≥5% in any treatment groups (VX-548, placebo, or HB/APAP, respectively) were nausea (19.0%, 25.2%, 32.8%), constipation (10.5%, 10.8%, 8.7%), headache (4.2%, 5.0%, 7.1%), dizziness (4.0%, 7.7%, 5.4%), and hypotension (2.5%, 6.8%, 3.6%).
VX-548 Phase 3 Results in Patients Undergoing Bunionectomy
Efficacy Results
Patients aged 18 to 80 years with moderate or severe pain after bunionectomy surgery were eligible to participate in the trial. 1,073 patients were randomized and dosed with either VX-548 administered orally with an initial dose of 100 mg followed by 50 mg every 12 hours (at 12, 24 and 36 hours after the first dose), hydrocodone bitartrate/acetaminophen (5 mg/325 mg administered orally every 6 hours over 42 hours) or placebo.
Primary and Key Secondary Outcomes in Phase 3 Study of Acute Pain Following Bunionectomy
Placebo
|
HB/APAP
|
VX-548
|
|
Primary endpoint of SPID48 vs. placebo |
|
|
|
LS mean SPID48 (SE) |
70.6 (6.3) |
-- |
99.9 (4.5) |
LS mean SPID48 difference from placebo |
-- |
-- |
29.3 |
95% CI |
-- |
-- |
(14.0, 44.6) |
P value vs. placebo |
-- |
-- |
0.0002 |
First key secondary endpoint of SPID48 vs. HB/APAP |
|
|
|
LS mean SPID48 (SE) |
-- |
120.1 (4.5) |
99.9 (4.5) |
LS mean SPID48 difference from HB/APAP |
-- |
-- |
-20.2 |
95% CI |
-- |
-- |
(-32.7, -7.7) |
P value vs. HB/APAP |
-- |
-- |
0.0016 |
Second key secondary endpoint of time to ≥2-point reduction in NPRS from baseline vs. placebo (minutes) |
|
|
|
Median time |
480 |
-- |
240 |
95% CI |
(476, 716) |
-- |
(117, 477) |
P value* vs. placebo (Log-rank) |
-- |
-- |
0.0016 |
*P value is nominal; CI = confidence interval; LS = least squares; SE = standard error. |
|||
Mean NPRS Over Time in Phase 3 Study of Acute Pain Following Bunionectomy [See Figure #2 at top of press release]
NPRS Reductions From Baseline at 48 Hours in Phase 3 Study of Acute Pain Following Bunionectomy
Placebo
|
HB/APAP
|
VX-548
|
||
Baseline NPRS, mean |
6.8 |
6.8 |
6.7 |
|
Change from baseline in NPRS at 48 hours, mean |
-2.6 |
-3.6 |
-3.4 |
|
% reduction from baseline in mean NPRS at 48 hours |
38% |
53% |
51% |
Safety Results
VX-548 was generally well tolerated in this study. The majority of AEs were mild to moderate, and there were no SAEs.
In general, AEs were consistent with the post-surgical setting. In the VX-548 arm, the incidence of AEs was lower than placebo (patients with any AEs in VX-548 and placebo arms: 31.0% and 35.2%, respectively).
AEs with an incidence ≥5% in any treatment groups (VX-548, placebo, or HB/APAP, respectively) were nausea (8.2%, 10.6%, 14.4%), headache (4.9%, 9.3%, 10.4%), constipation (3.5%, 4.2%, 5.1%), and dizziness (3.5%, 5.1%, 5.3%).
VX-548 Phase 3 Safety and Effectiveness Study Results
Vertex conducted an additional Phase 3 study in 256 patients to evaluate safety and effectiveness in a broad range of surgical and non-surgical moderate-to-severe acute pain conditions. Treatment with VX-548 administered orally with an initial dose of 100 mg followed by 50 mg every 12 hours for up to 14 days demonstrated safety, tolerability and effectiveness across these populations.
VX-548 was generally safe and well tolerated in this study. AEs were mostly mild or moderate, and there were no SAEs related to VX-548. The safety profile in this study was generally consistent with randomized, controlled Phase 3 studies with VX-548.
Patient perception of VX-548 effectiveness in treating pain as measured by a patient global assessment (PGA) at the end of treatment showed 83.2% of patients reporting VX-548 as good, very good, or excellent.
Next Steps for the Pain Portfolio
Vertex plans to submit a New Drug Application to the Food and Drug Administration (FDA) by mid-2024 with the goal of achieving a broad label in moderate-to-severe acute pain. VX-548 has secured Breakthrough Therapy and Fast Track designations in the U.S. for acute pain.
In addition, Vertex seeks to achieve a broad label in peripheral neuropathic pain. In support of this goal, the company recently reported positive Phase 2 results in painful diabetic peripheral neuropathy (DPN) and expects to advance to pivotal development in DPN with VX-548 following the end-of-phase 2 meeting with the FDA. Vertex has also initiated a Phase 2 peripheral neuropathic pain study of VX-548 in patients with painful lumbosacral radiculopathy, or LSR, which is pain caused by impairment or injury to nerve roots in the area of the lumbar spine.
In line with its portfolio strategy, Vertex continues to advance preclinical and clinical development of additional NaV1.8 and NaV1.7 inhibitors, for use alone or in combination, in acute and neuropathic pain.
Conference Call and Webcast
The company will host a conference call and webcast at 8:00 a.m. ET on January 30, 2024. To access the call, please dial (833) 630-2124 (U.S.) or +1 (412) 317-0651 (International) and reference the "Vertex Pharmaceuticals Conference Call."
The conference call will be webcast live and a link to the webcast can be accessed through Vertex's website at www.vrtx.com in the "Investors" section. To ensure a timely connection, it is recommended that participants register at least 15 minutes prior to the scheduled webcast. An archived webcast will be available on the company's website.
About Acute Pain
Defined as pain lasting <3 months, acute pain is a disabling condition that affects over 80 million people in the U.S. each year. Due to limited treatment options, there is an unmet need in acute pain management to improve the patient experience and reduce the economic and societal burden.
About VX-548
VX-548 is an investigational oral, selective NaV1.8 pain signal inhibitor that is highly selective for NaV1.8 relative to other NaV channels. NaV1.8 is a voltage-gated sodium channel that plays a critical role in pain signaling in the peripheral nervous system. NaV1.8 is a genetically validated target for the treatment of pain, and Vertex has previously demonstrated positive proof-of-concept results and a well-tolerated profile with VX-548 in two Phase 2 studies of acute pain following abdominoplasty and bunionectomy surgeries and in a Phase 2 study in painful diabetic peripheral neuropathy, a type of peripheral neuropathic pain. Vertex's approach is to selectively inhibit NaV1.8 using small molecules with the objective of creating a new class of medicines that have the potential to provide effective relief of pain without the limitations of opioids, including their addictive potential. VX-548 is one of the most recent molecules to enter clinical development from Vertex's portfolio of NaV1.8 pain signal inhibitors.
About Vertex
Vertex is a global biotechnology company that invests in scientific innovation to create transformative medicines for people with serious diseases. The company has approved medicines that treat the underlying causes of multiple chronic, life-shortening genetic diseases — cystic fibrosis, sickle cell disease and transfusion-dependent beta thalassemia — and continues to advance clinical and research programs in these diseases. Vertex also has a robust clinical pipeline of investigational therapies across a range of modalities in other serious diseases where it has deep insight into causal human biology, including APOL1-mediated kidney disease, acute and neuropathic pain, type 1 diabetes, myotonic dystrophy type 1 and alpha-1 antitrypsin deficiency.
Vertex was founded in 1989 and has its global headquarters in Boston, with international headquarters in London. Additionally, the company has research and development sites and commercial offices in North America, Europe, Australia, Latin America and the Middle East. Vertex is consistently recognized as one of the industry's top places to work, including 14 consecutive years on Science magazine's Top Employers list and one of Fortune's 100 Best Companies to Work For. For company updates and to learn more about Vertex's history of innovation, visit www.vrtx.com or follow us on LinkedIn, Facebook, Instagram, YouTube and Twitter/X.
Special Note Regarding Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended, including, without limitation, statements by Reshma Kewalramani, M.D., and Jessica Oswald, M.D., M.P.H., in this press release, and statements regarding our beliefs about the potential benefits of VX-548, our plans to submit a New Drug Application with the FDA by mid-2024, our goal of achieving a broad label in moderate to severe acute pain, our plans for a broad label in peripheral neuropathic pain, our plans to advance to pivotal development in DPN with VX-548 following the end-of-phase 2 meeting with the FDA, and our plans to continue to advance preclinical and clinical development of additional NaV1.8 and NaV1.7 inhibitors in acute and neuropathic pain. While Vertex believes the forward-looking statements contained in this press release are accurate, these forward-looking statements represent the company's beliefs only as of the date of this press release and there are a number of risks and uncertainties that could cause actual events or results to differ materially from those expressed or implied by such forward-looking statements. Those risks and uncertainties include, among other things, that regulatory submissions may not be completed on the anticipated timeline, or at all, that we may not be able to achieve a broad pain label for VX-548, that data from the company's research and development programs may not support registration or further development of its compounds due to safety, efficacy, and other risks listed under the heading "Risk Factors" in Vertex's most recent annual report and subsequent quarterly reports filed with the Securities and Exchange Commission at www.sec.gov and available through the company's website at www.vrtx.com. You should not place undue reliance on these statements, or the scientific data presented. Vertex disclaims any obligation to update the information contained in this press release as new information becomes available.
(VRTX-GEN)
View source version on businesswire.com: https://www.businesswire.com/news/home/20240129843577/en/